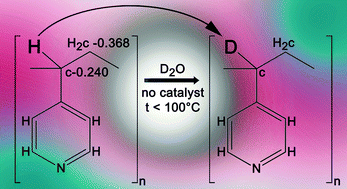
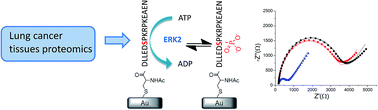
In this contribution we describe the formation of gold nanoparticles (AuNP) and polyaniline (PANI) AuNP-PANI nanocomposite via in-situ enzymatic polymerization. The method consists of electrostatic adsorption of anilinium monomers on AuNPs citrate stabilized surface of 50 nm diameters, followed by oxidation with horseradish peroxidase (HRP) enzyme and its cofactor H2O2. All reaction steps were monitored by UV-Vis-NIR spectroscopy including in-situ detection of the polymerization process. UV-Vis-NIR, Cyclic voltammetry (CV) and surface enhanced Raman scattering (SERS) measurements supported the formation of a nanoshell of PANI on the AuNP core. Two templates for anilinium assembly were compared revealing a strong dependence of the enzymatic kinetics on the template. The kinetic study had shown that the rigid template of the AuNP contributes to higher reaction rate on the AuNP compared with the more flexible polyanion template. The mild reaction’ condition enables an easy and precise method for obtaining PANI nano-shell on anionic templates for advanced bioelectronic applications.